Hace unos días tuve que
atender urgente en una guardia a una mujer de 49 años de edad que
venía derivada desde el ambulatorio. Había acudido allí por una
clínica de mareo próximo al sincope, de inicio brusco y reciente
mientras estaba trabajando, acompañado de temblores. En el
ambulatorio la encontraron algo "confusa" y decidieron
trasladarla al Servicio de Urgencias del Hospital para ampliar el
estudio. Entre los antecedentes médicos de la mujer solo destacaban
intolerancia a AAS, Asma bronquial alérgica, Espondiloartrosis y
ocasionales lumbalgias mecánicas y un tratamiento reciente con
Orgametril por una miomatosis uterina con metrorragias.
Clínicamente lo que a mí
me llamó la atención era que de entrada la mujer se mostraba algo
bradipsíquica, como torpe a pesar de manifestar un entendimiento y
juicio intactos y que presentaba unos movimientos que inicialmente me
hicieron pensar en una distonía oromandibular sin blefaroespasmo.
Había demás afectación
de los músculos superficiales del lado izquierdo del cuello, cosa
que tampoco es rara en estos casos.
Inmediatamente pensé en
un posible extrapiramidalismo iatrogénico dado el inicio brusco y le
pregunté a la paciente por el consumo reciente de nuevas
medicaciones como por ejemplo la metoclopramida, a la vez que
iniciaba un tratamiento de prueba con Akineton.
La sorpresa fue que en
breve, la paciente en vez de responder lo mas mínimo al tratamiento
comenzó a hacer episodios involuntarios de giro de la cabeza hacia
la izquierda y la cosa ya empezaba a parecer demasiado abigarrada
para ser una distonía. Era un cuadro que se iba haciendo complejo
por momentos, de inicio agudo y que por lo que parecía no tenía un
desencadenante tóxico o medicamentoso.
Decidí hacer
inmediatamente un TAC cerebral y minutos antes de llevarla al
servicio de radiodiagnóstico la paciente presentó movimientos
involuntarios en el MSI que me hicieron diagnosticar una crisis
comicial parcial. No había una progresión Jacksoniana clara de la
crisis.
¡Yo empezaba a tener
claro que la sintomatología de la paciente se debía a una lesión
en el SNC que irritaba el cortex cerebral!. No había datos clínicos
de HIC ni afectación de pares craneales por lo que imaginé además
que la sesión no producía mucho edema ni ocupaba espacio.
El TAC cerebral se hizo
inicialmente sin contraste y me llamo la atención que se podía
apreciar una lesión frontal derecha hiperdensa y con mínimo edema y
que no desplazaba la línea media cerebral. Estaba claro que esa
podía ser la causa de la clínica. Probablemente un tumor cerebral,
ya fuese este primario o metastasico, frontal y derecho que producía
una clínica de crisis parciales focales contra laterales por
irritación de la corteza cerebral.

Pero mirando con mas
detenimiento el TAC había algo que me desconcertaba y era un
borramiento de la línea media cerebral anterior y el difuminado del
contorno de las astas frontales de los ventrículos cerebrales,
aunque no podía decir que allí hubiese una lesión que ocupase
espacio, desplazase las estructuras anatómicas normales o
comprimiese los ventrículos. Parecía además como si hubiese en la
zona algo de hiperdensidad. ¡pero no se delimitaba nada!

Así que llamé al
radiólogo de guardia y realizamos un segundo TAC cerebral urgente
con contraste iv que inicialmente mostró lo mismo, aunque mejor:

Y que fue informado de la
siguiente manera por el radiólogo:
Lesión
espontáneamente hiperdensa de aproximadamente 1,5 cm a nivel frontal
que no realza significativamente tras administrar contraste y que se
acompaña de mínimo edema perilesional. Hallazgos compatibles con
tumoración primaria / metástasis
A mi me seguía llamando
la atención lo mismo que en el TAC sin contraste y estaba convencido
de que ahí había algo.


Se ingresó a la paciente y se le realizó una punción lumbar para analizar el líquido cefalorraquídeo que fue normal y pruebas de imagen destinadas a descartar una posible etiología metastásica de la lesión apreciada en el TAC (TAC toraco-abdomino-pélvico, mamografia bilateral y ecografía del cuello), que acabaron por no demostrar patología alguna de interés.
Una RMN cerebral ha
aportando posteriormente estas imágenes





Y estos cortes han
demostrando que yo no estaba equivocado.
¡Aquí está lo que yo
intuía!


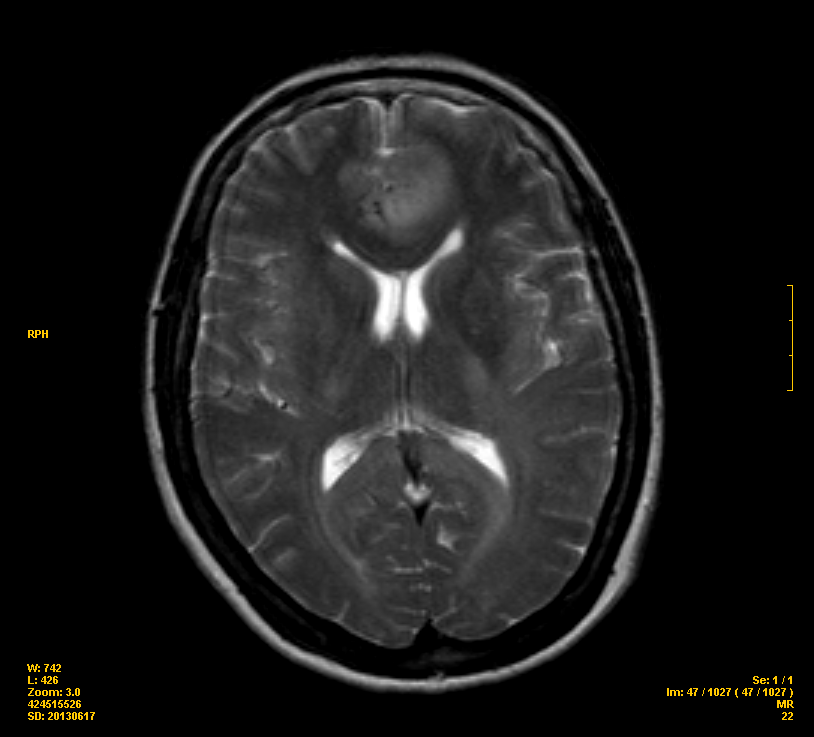

RMN cerebral con
realización de secuencias habituales, Difusión, Perfusión y 3D-T1
+Gd
Lesión infiltrativa
que presenta alta señal en T2 en sustancia blanca de ambos lóbulos
frontales, envolviendo el cortex, cruza la línea media,
extendiéndose a través de las fibras del cuerpo calloso. En el seno
de dicha lesión existen pequeños focos parcheados que realzan con
contraste en el lóbulo frontal izquierdo (giro cingular) y en
rodilla y tronco del cuerpo calloso. El componente izquierdo presenta
una discreta restricción de la Difusión y un ligero incremento de
la vascularización en los mapas rCBV (x 1-2). Sistema ventricular
centrado de calibre normal.
Además se vio que
En brazo posterior de
la cápsula interna izquierda y pedúnculo cerebral izquierdo existe
un área de alargamiento de la señal en T2 que no realza con Gd, de
similar significado.


Es muy llamativo lo poco
que se ve la lesión en el TAC y lo enorme que se aprecia en la RMN.
La explicación de eso es
que es una lesión que infiltra los tejidos anatómicos normales sin
provocar mucha distorsión de la estructura, efecto masa o edema.
Esta es una característica de algunos tumores cerebrales, en
particular de algunos Gliomas como el Glioblastoma. El caso infiltrativo por excelencia es
la Gliomatosis cerebri.
El glioblastoma (también conocido como glioblastoma multiforme) es el tumor más común y más maligno entre las neoplasias de la glía. Su nombre quedó€ establecido por la clasificación OMS-2000 y fijado por la clasificación OMS-2007. Existen dos variedades: el glioblastoma de células gigantes y el gliosarcoma.
Es un tumor de rápido crecimiento, compuesto por una mezcla heterogénea de células tumorales astrocitarias pobremente diferenciadas, con pleomorfismo, necrosis, proliferación vascular y frecuentes mitosis. Puede manifestarse a cualquier edad, pero afecta principalmente a adultos, con un pico de incidencia entre los 45 y los 70 años. Se presenta habitualmente en los hemisferios cerebrales, siendo menos frecuente su localización en el tronco del encéfalo o la médula espinal. Al igual que todos los tumores cerebrales, excepto en casos muy raros, no se expande más allá de las estructuras del sistema nervioso central. Puede desarrollarse a partir de un astrocitoma difuso (grado II) o de un astrocitoma anaplásico (grado III) (en tal caso se denomina secundario), pero con mayor frecuencia ocurre de novo, sin ninguna evidencia de neoplasia previa (denominándose en este caso primario). Si bien el glioblastoma es el tumor cerebral primario mas frecuente, su incidencia es de sólo 2-3 casos por cada 100.000 personas en Europa y Norteamérica.
La historia clínica de la enfermedad suele ser corta (menos de 3 meses, en mas del 50% de los casos), a menos que el tumor no se desarrolle por la progresión de un astrocitoma de bajo grado (glioblastoma secundario).
Los síntomas del glioblastoma son los de una masa expansiva en el interior del cráneo, que aumenta la presión intracraneal. Es común por tanto encontrar cefalea, nauseas, vómitos, dilatación de los vasos cerebrales con alteraciones de la retina hasta el papiledema, hemiparesia, hemianestesia, hemianopsia, diplopia, afasia y crisis convulsivas. El porcentaje de los pacientes que experimentan ataques epilépticos asciende a un tercio. Destacan también síntomas neurológicos no específicos tales como el obnubilamiento de la conciencia y los cambios de personalidad.
En el tratamiento del glioblastoma intervienen la cirugía, la radioterapia y la quimioterapia. A pesar de todo el arsenal terapéutico su pronóstico es infausto, con una mediana de supervivencia de aproximadamente 14 meses. Son raros los casos de supervivencia prolongada, aunque se han descrito.
El glioblastoma (también conocido como glioblastoma multiforme) es el tumor más común y más maligno entre las neoplasias de la glía. Su nombre quedó€ establecido por la clasificación OMS-2000 y fijado por la clasificación OMS-2007. Existen dos variedades: el glioblastoma de células gigantes y el gliosarcoma.
Es un tumor de rápido crecimiento, compuesto por una mezcla heterogénea de células tumorales astrocitarias pobremente diferenciadas, con pleomorfismo, necrosis, proliferación vascular y frecuentes mitosis. Puede manifestarse a cualquier edad, pero afecta principalmente a adultos, con un pico de incidencia entre los 45 y los 70 años. Se presenta habitualmente en los hemisferios cerebrales, siendo menos frecuente su localización en el tronco del encéfalo o la médula espinal. Al igual que todos los tumores cerebrales, excepto en casos muy raros, no se expande más allá de las estructuras del sistema nervioso central. Puede desarrollarse a partir de un astrocitoma difuso (grado II) o de un astrocitoma anaplásico (grado III) (en tal caso se denomina secundario), pero con mayor frecuencia ocurre de novo, sin ninguna evidencia de neoplasia previa (denominándose en este caso primario). Si bien el glioblastoma es el tumor cerebral primario mas frecuente, su incidencia es de sólo 2-3 casos por cada 100.000 personas en Europa y Norteamérica.
La historia clínica de la enfermedad suele ser corta (menos de 3 meses, en mas del 50% de los casos), a menos que el tumor no se desarrolle por la progresión de un astrocitoma de bajo grado (glioblastoma secundario).
Los síntomas del glioblastoma son los de una masa expansiva en el interior del cráneo, que aumenta la presión intracraneal. Es común por tanto encontrar cefalea, nauseas, vómitos, dilatación de los vasos cerebrales con alteraciones de la retina hasta el papiledema, hemiparesia, hemianestesia, hemianopsia, diplopia, afasia y crisis convulsivas. El porcentaje de los pacientes que experimentan ataques epilépticos asciende a un tercio. Destacan también síntomas neurológicos no específicos tales como el obnubilamiento de la conciencia y los cambios de personalidad.
En el tratamiento del glioblastoma intervienen la cirugía, la radioterapia y la quimioterapia. A pesar de todo el arsenal terapéutico su pronóstico es infausto, con una mediana de supervivencia de aproximadamente 14 meses. Son raros los casos de supervivencia prolongada, aunque se han descrito.
La Gliomatosis cerebral
(Gliomatisis cerebri) es una rara neoplasia infiltrante
(aproximadamente el 1% de todos los tumores cerebrales) de origen
glial descrita inicialmente por Nervin en 1938 (Nevin S. Gliomatosis
cerebri. Brain 1938; 61: 170-91) y que afecta al cerebro con un
sobrecrecimiento difuso del mismo y preservación de las estructuras
neuronales subyacetes. Las
células gliales neoplásicas infiltran fundamentalmente a través de
la sustancia blanca, afectando y expandiendo con frecuencia
estructuras de conexión interhemisféricas como
el cuerpo calloso, pudiendo extenderse también hacia la médula
espinal. Es
reconocida por la clasificación de tumores cerebrales de la
Organización Mundial de la Salud (OMS) desde 1979 como una entidad
específica entre los tumores neuroepiteliales de origen incierto. De
acuerdo con los criterios de la World Health Organization (WHO)
Classifcation
of Tumours, debe
afectar a un mínimo de 3 lóbulos cerebrales y puede infiltrar
sustancia gris tanto cortical como de ganglios basales, así como
extenderse a lo largo del tronco cerebral y la médula espinal. El
fenotipo celular habitualmente es de tipo astrocitario aunque también
puede
ser
oligodendrocitario u
oligoastrocitario. Se ha dividido en
2 tipos:
Tipo
1, cuando no se observa una masa tumoral en el patrón infilltrativo,
Tipo
2, cuando además de la infiltración cerebral se objetiva una lesión
con efecto de masa, habitualmente pequeña (< 1 cm de diámetro).
Cuando
otros tipos de gliomas presentan una extensa progresión de
características infiltrativas,
más que crecimiento de la masa tumoral en sí misma, se denomina a
estos fenómenos gliomatosis cerebral secundaria. La gliomatosis cerebral presenta un comportamiento
biológico habitualmente agresivo, por lo que la WHO la clasifica
como grado III de malignidad.
Es
más frecuente en las tercera y quita décadas de la vida aunque
puede presentarse a cualquier
edad y sin predilección por sexo.
El diagnóstico clínico
es difícil ya que la presentación clínica es muy variable
dependiendo
del área del sistema nervioso central comprometida. Las
manifestaciones más frecuentemente halladas son crisis
comiciales, alteraciones cognitivas,
déficit focales y cefalea. Es típico de estos
pacientes
una
clara disociación clínico radiológica, de manera que la
extensión de la lesión por neuroimagen
es mayor de lo que refleja la repercusión clínica.
La enfermedad suele ser
de progresión lenta. El TAC no presenta hallazgos típicos siendo
mucho mas sensible en su detección la RNM que muestra mejor la
extensión del tumor (sobre todo en las secuencias potenciadas en
T2), pudiéndose sugerir el diagnóstico cuando se detecta una lesión
cerebral difusa, sin masa ni necrosis, que produce un sobre
crecimiento de estructuras cerebrales con tendencia a afectar vías
de conexión de la línea media.
El diagnóstico
diferencial por imagen incluye a otros gliomas infiltrantes como los
Astrocitomas anaplásicos y los oligodendrogliomas que suelen
presentarse como masas y realzan de forma mas significativa tras la
administración del contraste paramagnético. Otras entidades con las
que hay que hacer el diagnóstico diferencial de imagen son el
linfoma cerebral difuso enfermedades desmielinizantes como la esclerosis múltiple y la
leucoencefalopatia multifocal progresiva.
El
diagnóstico definitivo se realiza mediante estudio
anatomopatológico de material de biopsia. Hay casos publicados en la
literatura en los que se realiza la aproximación diagnóstica
mediante manifestaciones clínicas y neuroimágenes y en los que posteriormente el diagnóstico se confirmaba en el examen post
mortem.
La
supervivencia media es de alrededor de 12 meses desde el diagnóstico.
¡Sic!
Por cierto, para los que
les interese, la última versión de la clasificación de los tumores
del SNC es el fruto de un consenso entre 25 patólogos y genetistas
celebrada en el Centro Alemán para la Investigación del Cáncer, en
Heidelberg a finales del 2006 y está recogido e un fascículo de la
Organización Mundial de la Salud (OMS): Louis DN, Ohgaki H, WiestlerOD, Cavenee WK (eds) (2007). WHO Classification of tumours of theCentral Nervous System. IARC, Lyon.
La clasificación es la
misma de la edición anterior (2000) a la cual se añaden nuevas
entidades descritas o redefinidas en los siguientes 6 años.





Finalmente la biopsia estereotáxica del paciente de este caso aportó el diagnóstico de GLIOBLASTOMA, GRADO IV DE LA OMS. El estudió inmunohistoquímico demostró una inmunorreactividad para PGFA, EGFR y p53 Y el índice de proliferación celular medio con ki67 fue muy alto.
Extremadamente interesante este caso clínico y muy biem. Explicado.
ResponderEliminarMuy biem explicado y muy interesante Juan
Quizá pueda interesarle, o al menos, ayudar a dar la difusión que el proyecto de investigación requiere.
ResponderEliminarGracias y un saludo
http://izaslaprincesaguisante.org/investigacion-2/proyecto-investigacion-en-curso-hospital-san-juan-de-dios-barcelona/